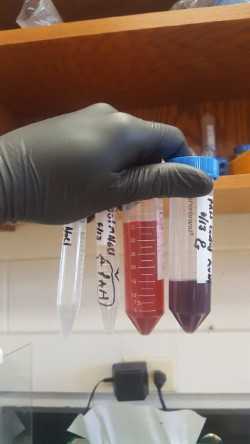
So last week Monday morning came around and Professor Thompson had been too busy to prepare gold nano-particles for me. This was fine, I was ready to tackle the task on my own. The procedure involves bringing 100mL of a solution up to a boil and then pouring 3mL of another solution into it. The solution then proceeds to change colors several times, its actually pretty neat to watch. I ended up making a pretty concentrated batch of nano-particles. It even ended looking pretty good under the UV-Vis, so I decided to continue on with my project using them. I needed to find a good ratio of NPs and PAH because the two are pretty fragile. If not mixed together properly they can immediately aggregate and make the solution unusable. Also, if they are not in the proper ratio this can also cause it to become aggregated and then unusable. So I tried to make a 3x and a 5x diluted NPs solution with normal concentrations of PAH. Both seemed usable after mixing. I then tried out the new cleaning procedure that Professor Thompson had suggested. I would spin the solutions at very high speeds, 9000 rxg and 7000 rxg in order to get all of the excess PAH out of the solution. This maximized our risk of contamination by stray PAH particles. My problem with this procedure arose when I had to wash the pellets with DNA in them. After I started this centrifugal process, I found that the solutions were creating pellets that were virtually non-existent. This is not good, as it is one of our main goals to get good pellets here. I was thinking that these pellets could not form because I had diluted the initial amount of NPs in the beginning. Professor Thompson suggested it had something to do with my spinning procedure for the DNA washes. I’m now currently trying this new procedure, which includes longer times and a larger rxg, to get better pellets in my solutions.
No comments