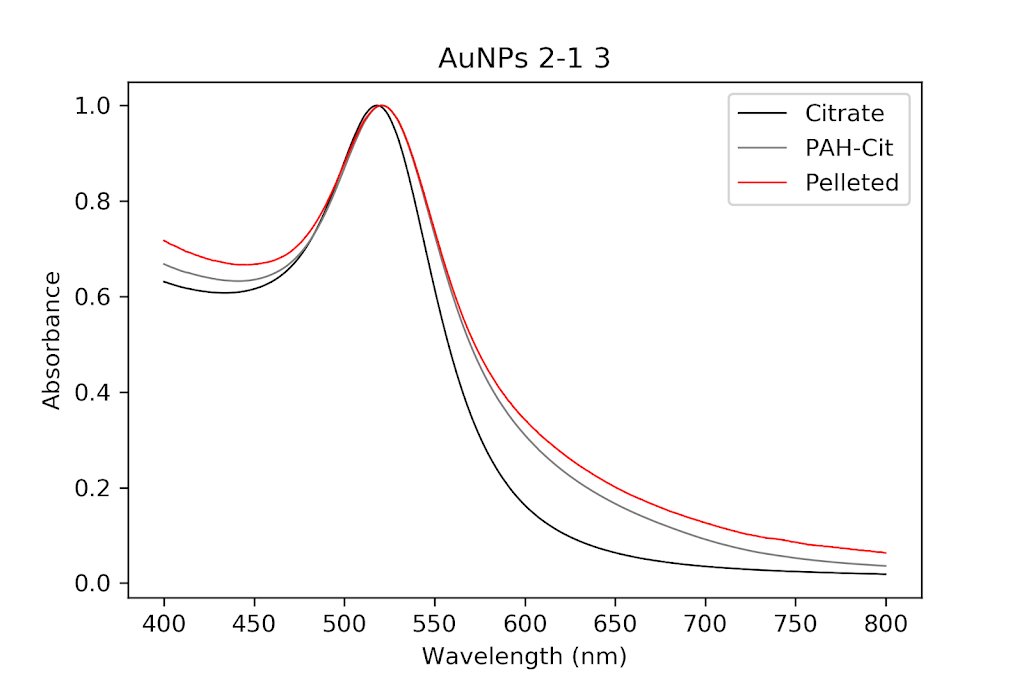
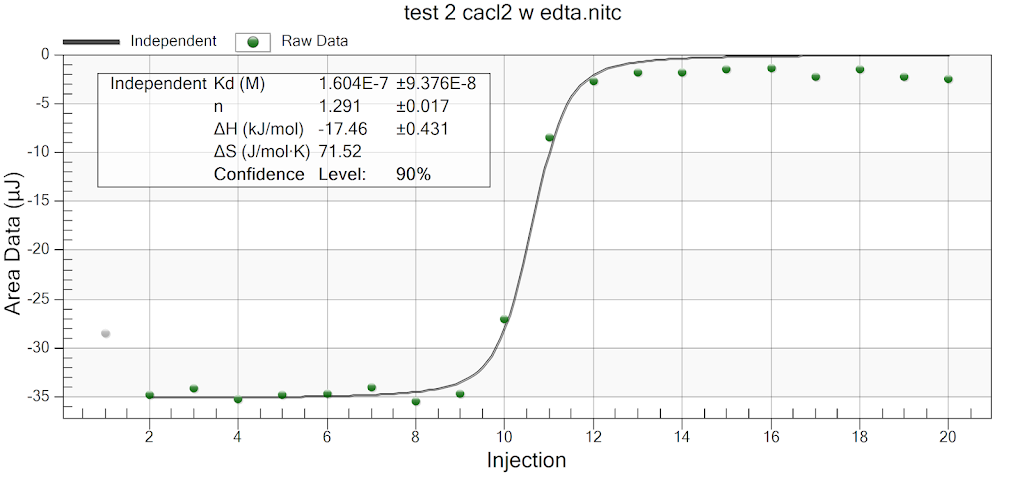
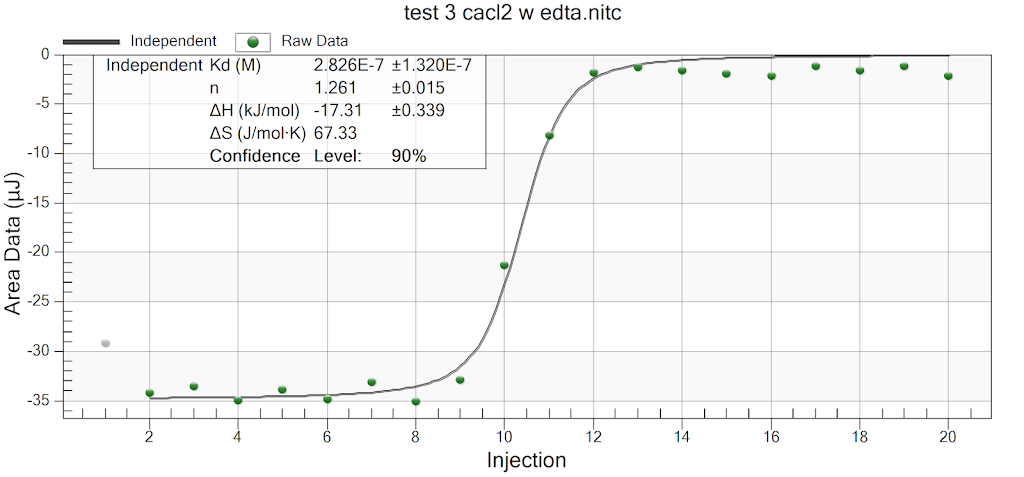
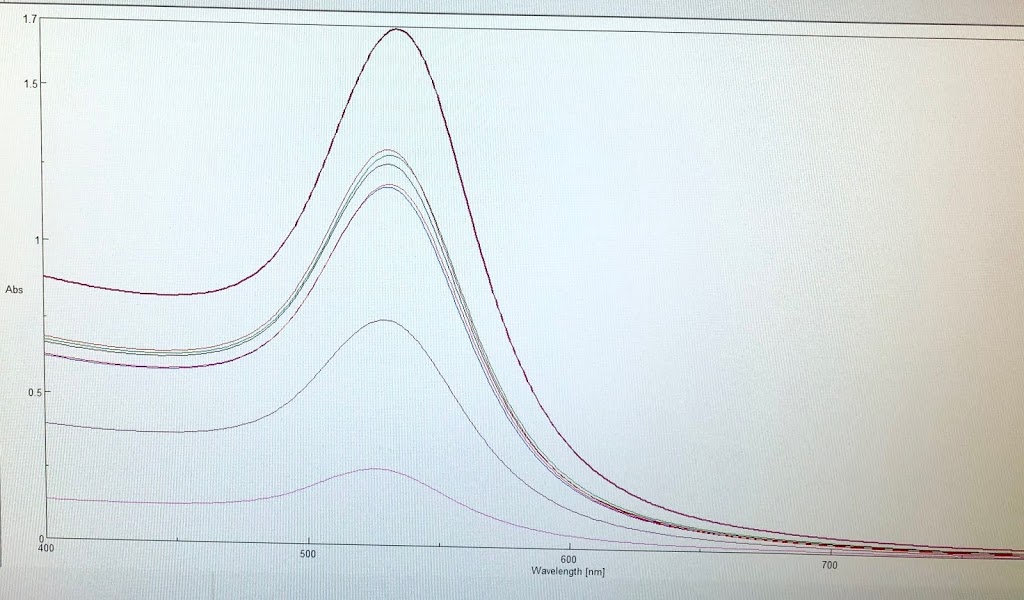
During the beginning of these weeks, I focused on perfecting the polymer coating procedure from before. I got a few good attempts, in which the data looks decent enough to move on to DNA wrapping, but overall, there is no deciding procedure that works the best. This is somewhat disappointing; when I need to make new PAH-Cit AuNPs in the future, I have to either get lucky or find a way that does work most of the time. For now, however, I am going to use the ones that are decent enough to move forward. These are all from the same batch but are separated into 15mL conical tubes in order to make the centrifugation go smoothly. Their absorption graphs look decent, with only a small shoulder which indicates some aggregation of particles. This isn’t too big of an issue, though, so they are cleared to wrap with DNA.
In order to get the particles ready for DNA wrapping, I needed to make the concentration higher. The concentrations were extremely low which would not work very well. To do this, I centrifuged them at 4000rpm for 45 minutes, removed the supernatant on top, combined the pellets and recentrifuged the supernatant until no pellet emerged. This would eventually give me a concentrated sample of NPs.
I also needed to prep the DNA for wrapping. To do this, I made a sample of DNA dissolved in TRIS buffer that has a steady PH. Once dissolved, I sheared the sample with a probe sonicator. This is a technique that essentially shortens the DNA to a specific length that allows for wrapping. The machine makes a really loud noise, so I had to wear ear protection. After this, I realized I used the wrong concentration of TRIS buffer before, so I had to dilute it in ultrapure water (deionized to ensure the DNA’s charge was not messed with) and a certain concentration of NaCl, which is needed later in the process to ensure the DNA and polymer are all surrounded with charges, but we decided to add it in then to save time. I also measured the absorption spectrum of both the sheared and unsheared DNA samples, in order to obtain their concentrations. This didn’t turn out well.
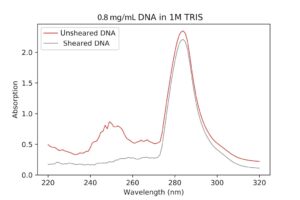 |
| Sheared DNA VS Unsheared DNA UV-Vis spectroscopy. The peak should be at 260nm, but it’s much higher. I’m not really sure why this is, although it might have to do with the machine not being calibrated correctly or my TRIS concentration being too high. |
Once the sheared DNA had been buffer exchanged, I ran another UV-Vis characterization to see if they were any better. I then realized I had been using a plastic cuvette to measure the spectrum, which, if you know anything about physics, is impenetrable by UV rays. Since the wavelength parameters are set to numbers in the ultraviolet range, I wasn’t actually getting real data. So, I re-ran the measurement with a quartz one, and they turned out good, with peaks of about 260nm (expected).
The absorption value at the peak, 0.13587, enabled me to calculate the concentration of DNA in the solution, which turned out to be around 0.68 mg/mL. For wrapping, I need to dilute this by about 10x.
Meanwhile, I finished pelleting my particles and needed to calculate their concentration with the UV-Vis as well in order to start wrapping.
 |
| Extremely concentrated gold nanoparticles. This is only about 2mL, as each time the pellets are combined it only adds a few μL of particles. These were already cleaned with the centrifuge process before pelleting. |
When I ran the UV-Vis I had to dilute the pellet 100x. The particles looked decent. Some mild aggregation and thickening of the peak had occurred, but I deemed this not significant enough to not continue wrapping. In python, I programmed the files from the machine in order to set up a graph, as usual. However, this time, in order to clean the samples, I needed to separate them into smaller tubes, 8 to be exact. There were 8 samples that looked extremely similar to one another and were going to be recombined anyway. To get the picture that you see below, I attempted to take the mean of the absorption values of all 8 samples at each wavelength, normalize them, and plot them again as a new line. This took me two hours, but it was worth it, as now the graph is much cleaner looking and easier to understand.
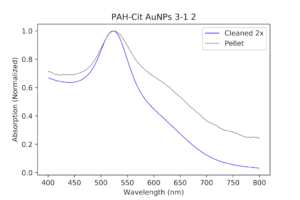 |
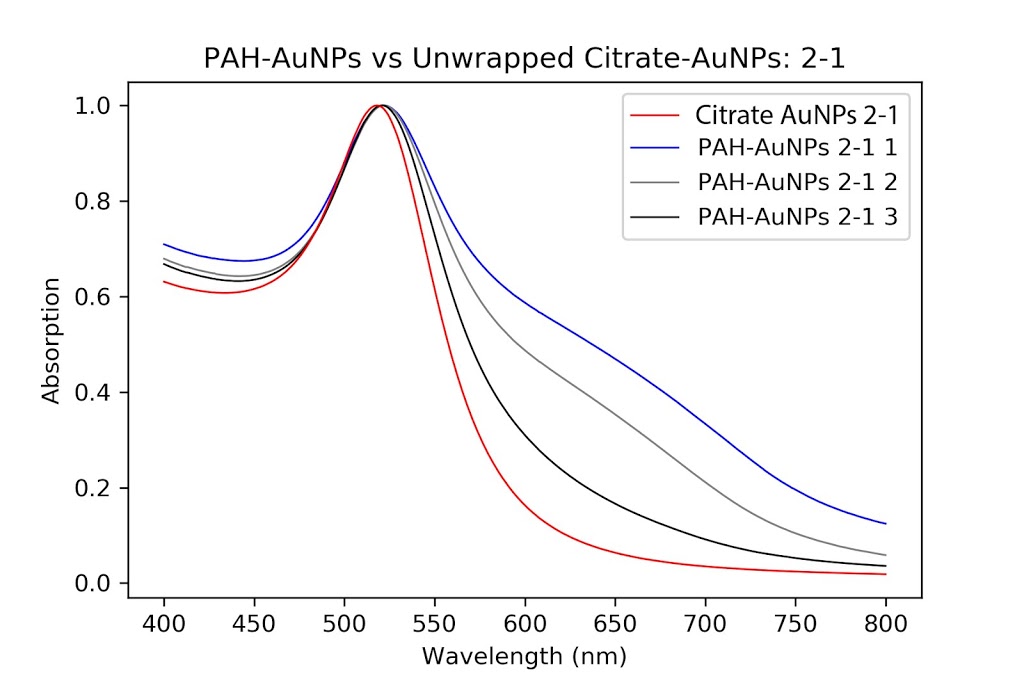
| The original PAH-Cit AuNPs cleaned twice with a centrifuge versus the pellet solution (a dilute version), normalized. As you can see, the pellet has a thicker peak and a slightly bigger shoulder. |
With this data, I used the Wolfgang Haiss protocol from another academic paper in order to determine the concentration of the particles. The procedure is as follows: take the absorption value at the highest peak, divide it by the absorption at 450nm, use the table to determine the diameter, and then use another table to determine the molar decadic extinction coefficient, which can then be used in the equation c = A450/ε450 in order to determine the concentration in moles. All of the calculations have been done in the paper, so I simply had to follow the protocol. When I did this, I finally got a concentration of 39nM for my pellet. I then diluted it by 10 in order to not overwhelm the DNA.
 |
| The first ingredient for the final mixture, diluted pellet from my PAH AuNPs. |
I diluted the DNA 10x simply by combining 1mL with the particles instead of diluting it with water and then adding 10mL, which is essential because I don’t want the water to contaminate the concentrations of NaCl and TRIS that the DNA is combined with. This gives me a final concentration of 0.068mg/mL DNA.
 |
| The second ingredient for the final mixture, diluted sheared DNA. |
This is my first attempt at creating DNA-PAH-Cit coated AuNPs. I shook it vigorously in order to make the DNA wrap around the particles, which Dr. Andresen said might work because we really have no idea what we’re doing and we have to try something. I then left it overnight.
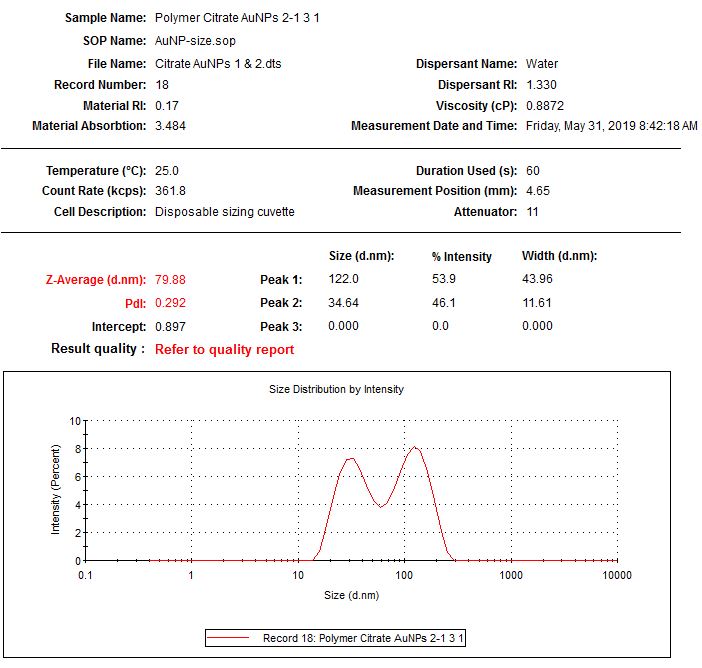
The next morning, they were a little purple in color. I ran UV-Vis measurements as well as DLS and Zeta potential. The absorption spectrum looked worse than the pellet, which I guess is to be expected, but since the pellet was already kind of aggregated, it made it worse.
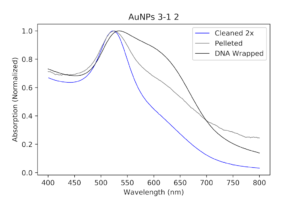 |
| My DNA wrapped particles compared to the original cleaned and pelleted polymer particles. The black line, indicative of the DNA wrapping, is aggregated. Ugh. |
I still wasn’t completely sure if this was terrible or not, so I began to clean the wrapped particles 2x in the centrifuge and run the tests again to see if they were better. After cleaning, I noticed that the tube had a clump of aggregated particles at the bottom and I immediately knew they weren’t going to be useable, but I still ran them through the UV-Vis, confirming my doubts. So much for the first attempt.
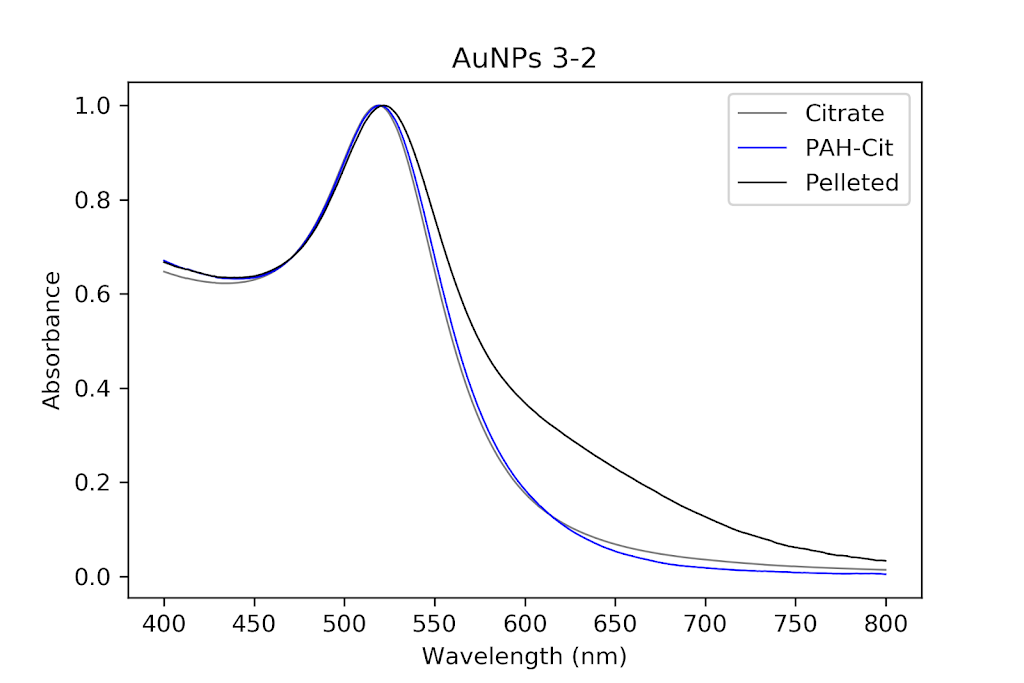
I decided to redo wrapping, but with a different set of particles. Luckily, I had other even better PAH ones that I had been pelleting along with the others and eventually worked my way up to 1mL of pellet. I then checked their absorption spectrum to ensure these were still as good as they were before. These particles were so much better it was insane. There was almost no shouldering even on the pellet solution, as opposed to my first try.
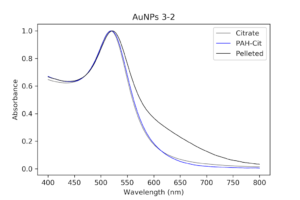 |
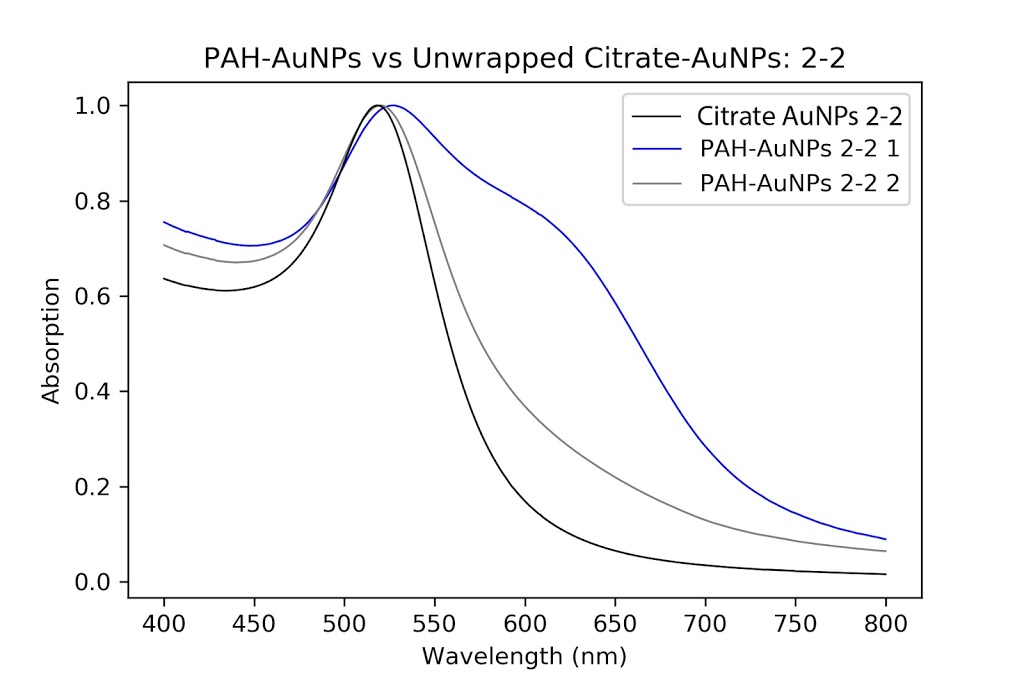
| Pelleted particles compared to original citrate and PAH-coated particles. As indicated by the black line, there is almost no aggregation and only slight broadening of the peak that has occurred. This has potential. |
I knew I had to save as much of this pellet as possible and not waste it with possible aggregation with DNA each attempt. I decided to dilute the pellet 50x instead of 10x in order to save the particles for as many tries as possible. I made 3 samples of the diluted pellet because the pellet has a greater chance of aggregating on its own if let sit out, simply because the particles are more concentrated and closer together. I only used 200uL for each dilution, which is great. I calculated the concentration using the method from before, and then I mixed the two ingredients together, 1mL sheared DNA from before and 9mL of the diluted pellet, in order to make 10mL of the DNA wrapped particles. Instead of shaking it vigorously this time, I decided to invert the tube a few times and let it sit a while instead. I noticed that I did the same thing when coating with PAH, and even when I made the original citrate particles. I didn’t disturb them at all, I just mixed them a little and then let them sit. I was hopeful that this would not cause aggregation but would still allow for the DNA to wrap around the particles.
 |
| My DNA-PAH-Cit AuNPs attempt 2. As you can see, they are a nice pink color, as opposed to the purplish color that arose from the previous try. These should be better. |
I then let them sit for a few hours and ran UV-Vis and DLS/Zeta on them. They seemed a little weird – the UV-Vis looked somehow unchanged from the dilute pellet, which was strange because I was expecting at least a little aggregation. The zeta potential was also weird because the surface charge was about 40mV. I was expecting this number to be negative since DNA is negatively charged, so I guess wrapping didn’t occur. The pellet’s charge was about 55mV, so I guess it did reduce it a little, but not nearly enough. It should be around -30mV. I deduced two things from this. Either the wrapping didn’t occur because I didn’t shake it enough (maybe the particles are stubborn), or I didn’t add a high enough concentration of DNA, so not enough stuck to the particles. I’m not sure what concentration to do next, or if I should stick with the original and try to shake it a little more. I’ll do that next week.
Finally, I started pelleting my other particles that were good, even though they are only 10mL each. Next week, I’ll finish pelleting these for later on, as well as make a new batch of DNA wrapped particles probably using a different concentration. I’m just happy that I was able to progress to the final step of preparation in my research. After I get these and they look good, I can begin experimentation. Hopefully, they work out!