On Monday, Dr. Andresen and I began the process of preparing nucleosomes. We began with 50mL of chicken blood which if you’re wondering the reason behind why we use chicken blood, it’s because unlike our red blood cells, chicken red blood cells have nuclei that contain nucleosomes.
First off, Dr. Andresen and I began the process of preparing nuclei. We made 500mL of KTM buffer, which contains Tris HCL, KCl, MgCl2 and PMSF, and combined 50mL of it with the 50mL of chicken blood. Then we spinned that using a Centrifuge machine. The Centrifuge machine separates the heavier stuff (the pellet) from the lighter stuff (the supernatant) by increasing the gravitational force.Therefore, for the first couple of spins it helped us separate the red blood cells (the pellet) from the extra stuff that the blood contains such as plasma (the supernatant). Then, we resuspended the cells in KTM buffer and Triton X. The Triton X is like a “soap” which helps “break” the cells in order to release the nuclei inside of them. We had to do this because we want what’s inside of nuclei, the nucleosomes.
Tuesday, we had a discussion about unproductive and productive stupidity in STEM. Right after the discussion, I actually made a mistake and added too much of one solution to a batch of KTM buffer that I needed to use in order to finalize the preparation of the nuclei. Dr. Andresen realized that I did this once he noticed how hard the pellet (which contained the nuclei) was since it wouldn’t dissolve in the KTM buffer. Therefore, I had to start all over. I was frustrated at the fact that I had ruined a day of work but it actually helped me get over the fear that I had of messing up. It began my journey of understanding that research is a process of trial and error and that my mistakes will only help me learn and grow.
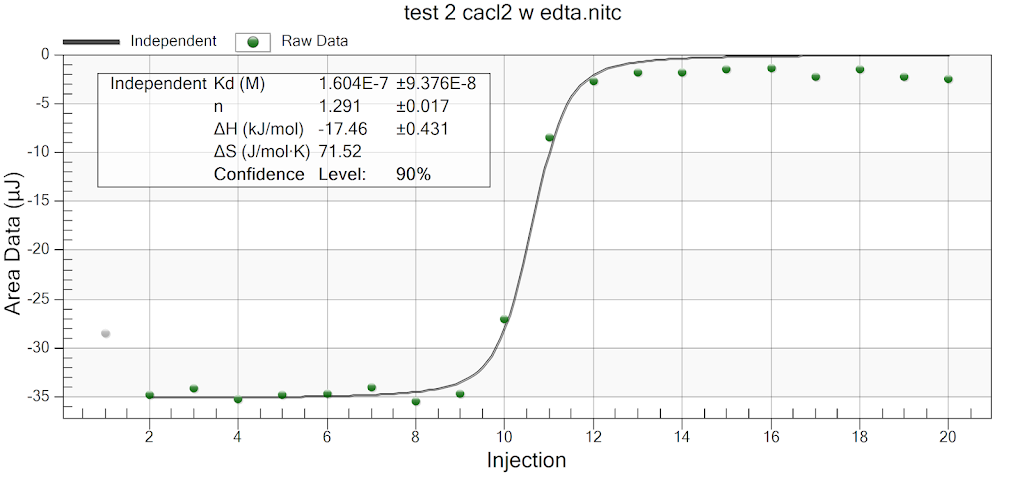
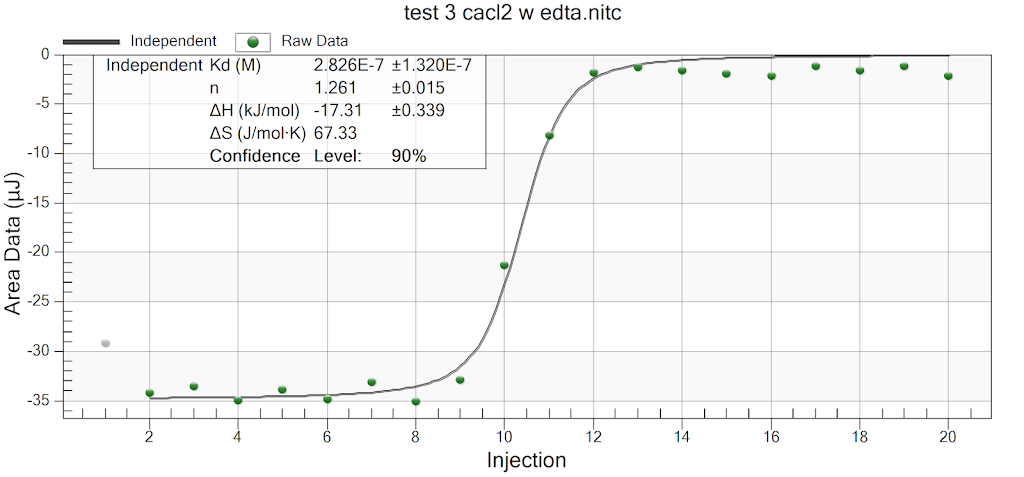
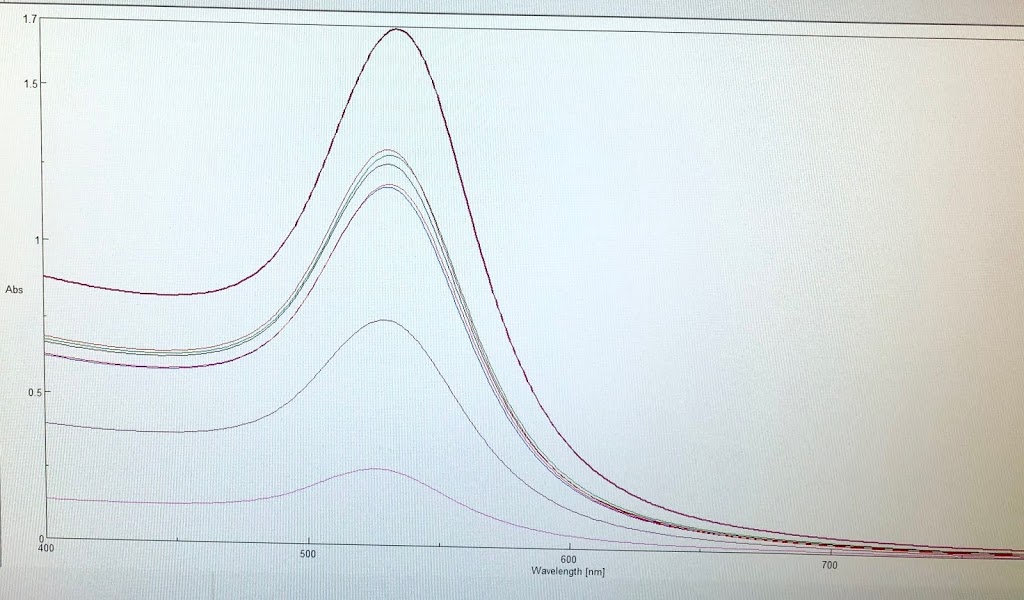
For the remainder of the week, Dr. Andresen and I went from having nuclei to having chromatin. When we had the nuclei, we measured how much DNA we had using the UV/VIS machine, which told us the concentration of DNA we had. We had about 125mg of DNA. Then, we resuspended the nuclei with CaCl2 + ML buffer, which contains Tris HCl, NaCl, MgCl2 and PMSF, and heated it at 37 degrees Celsius for 35 minutes. We heated this because throughout the process, the solution actually eats at the membrane of the nuclei which then breaks the nuclei and releases the DNA and everything else in the nuclei. Then, we continued by doing different steps in order to get the chromatin which consisted of making other solutions, spinning it, and letting it sit overnight with different solutions in order to get rid of the extra stuff such as ions and molecules surrounding the DNA.
As of today, Friday, we have chromatin, which consists of DNA linking nucleosomes together. Ultimately, what we’re trying to do is get the nucleosomes, which is an octamer wrapped around twice by DNA, so we’ll have to cut the DNA linking the nucleosomes in order to get the nucleosomes alone. Also, today we measured how much DNA we have, which is approximately how much chromatin we have. We have 37mg of DNA which is about 30% of how much we had last time we had checked. We are hoping to at least have half of this amount by the end. However, during our group meeting today I found out that the next step is where things have gone wrong in previous years. Even so, I am hoping to have some nucleosomes next week.