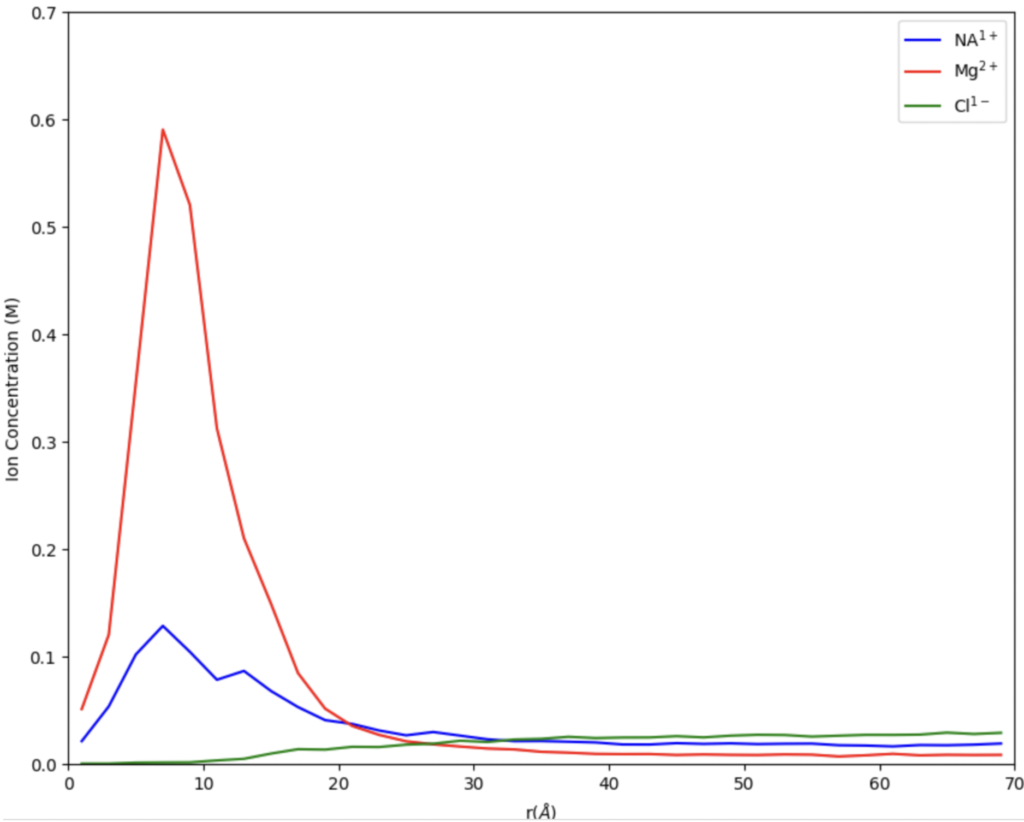
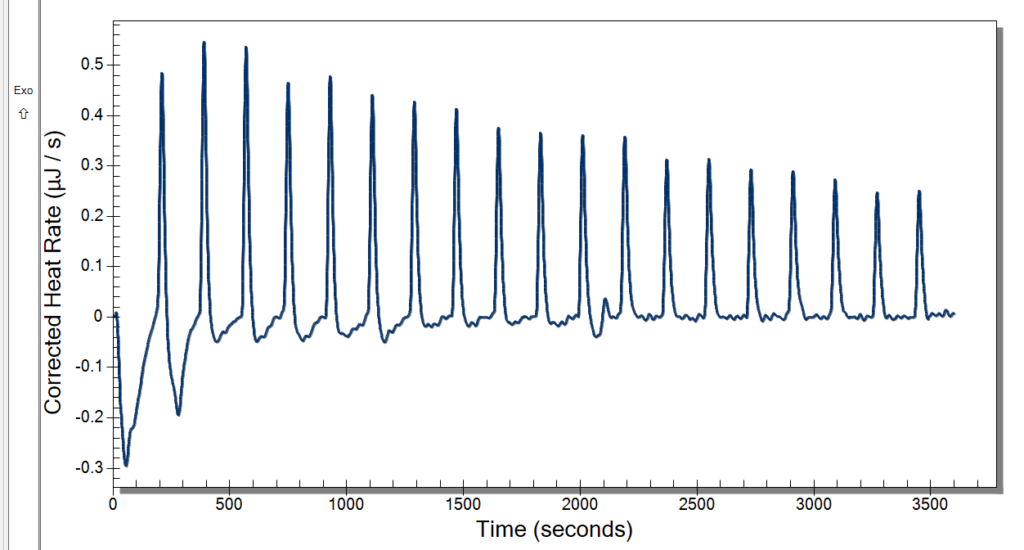
When we first started out our research this summer we were working on finding the best digestion for our chromatin. Digestion, simply put, is a way to get a fragment of a larger substance, in our circumstance, we are digesting chromatin to get nucleosomes. Our Senior Investigative Researcher (SIR) Macyn Rosay helped us to begin this process and slowly got us to do the process on our own. For the past week, we have been doing the digestion independently, looking for the best time for the 15-unit digestion. In the prior week, we had done a unit digestion that led us to 15 units the best for the digestion of our chromatin (so many gels). Unit digestion is how to find the baseline of Micrococcal Nuclease (MCN) to get the most amount of nucleosomes. From there we did a time digestion that ranged from 10 minutes to 55 minutes, and from this digestion, we concluded 30 minutes was the best for nucleosomes (see Fig. 1). Since 30 minutes was our blessed digestion we had (finally) moved to the next step which is mass digestion (only one more gel yay). The digestion process will be explained more in-depth later, for now, enjoy our fluorescent gel.
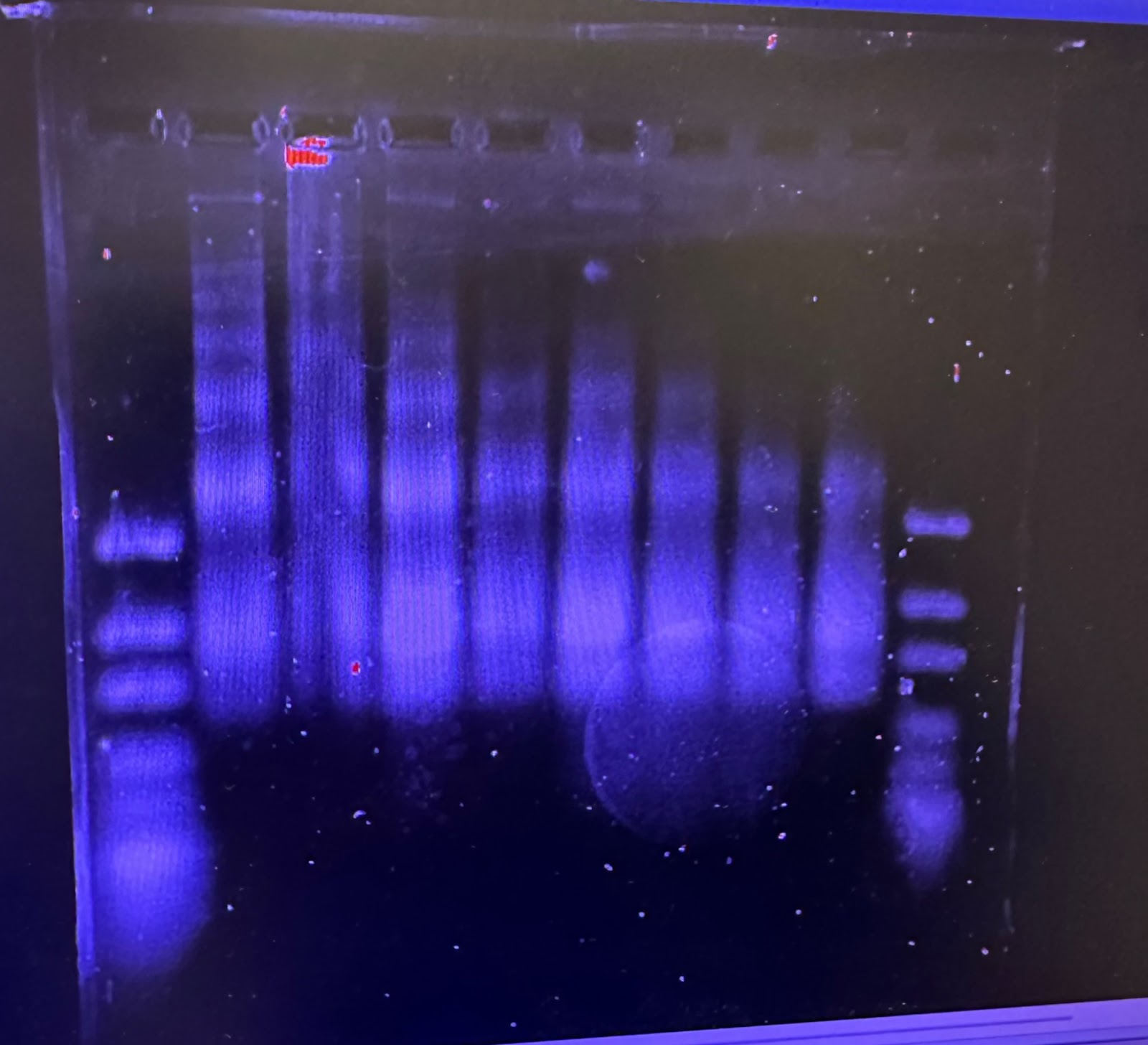
Figure 1: 15 unit Time Digestion of Chromatin
Once we (finally) made a good gel, we could move on. We spent the rest of the day digesting chromatin. We had a sample of 50 mL of chromatin that we separated into two separate test tubes with 25 mL in each just to make sure if we mess up (which we won’t), we are not using up all of the chromatin that we already have. If we do, then its back to the farm to get more chicken blood!
We then added 4.5 mL of MCN to each of the two test tubes filled with chromatin and then centrifuged them. After that, we equally separated this new solution (chromatin + MCN) into 5 test tubes with 5 mL each and heated these samples for 30 minutes at 37 ℃. It was now time to make (another!) gel (and listen to Lana Del Rey of course). Once 30 minutes had passed, we combined the 5 samples and added 1.25 mL of EDTA before putting it on ice for about 10 minutes (time for more Lana).
Next, we prepared two samples for the gel, one with 5 μL of chromatin, 5 μL proteinase K, and 0.5 μL SDS. The other sample had 5 μL of digested chromatin rather than the undigested chromatin. We then placed these two samples into the isotemp for 50 minutes at 50 ℃.
It was now time to move on to a higher-power centrifuge, which Professor Andresen taught us how to use. We added the digested chromatin to two centrifugal filter units and let those spin for about 10 minutes. In the meantime, we made a sample of 1.0 M NaCl which we later used to make 1.0 L of TEM buffer (Tris, EDTA, NaCl).
We then finished the digestion and loaded the gel before letting it run for about 2.5 hours. While it ran we took a lunch break at the world-renowned Bullet Hole (it is THAT good). Once they were ready, we analyzed the gels and got the thumbs up from Professor Andresen (YAY).
Figure 2: Digestion of Original Chromatin & 30 min Digested Chromatin
So through some rough patches in the first few weeks, we made a breakthrough, but even when we were going insane we always had great music playing.
Signing Off
JIM 2 (Eden) and JIM 3 (Kyle)